The Evolution of Osteogenesis Imperfecta through the Lens of Four Generations
- Aug 18, 2024
- 20 min read
Adapted from a presentation by Jane O'Hara at the Sonography Canada conference in October 2024.

The profile depicted above illustrates the ultrasound image of an infant diagnosed with Osteogenesis Imperfecta (OI). This child represents the fourth consecutive generation affected by this condition, following her father, grandmother, and great-grandmother. The family's willingness to share their experiences provides an invaluable case study, offering insights into the complexities of living with OI across multiple generations. This includes the challenges of raising a child with OI, the developmental experiences of individuals with OI, and the considerations involved in managing subsequent pregnancies in light of contemporary medical advancements and treatment options.
Osteogenesis Imperfecta, colloquially known as "brittle bone disease," is characterized by a propensity for fractures, particularly in the long bones, due to a defect in collagen production. The rare condition occurs in approximately 1 in 15,000 to 20,000 births. Given its rarity, many medical professionals, including sonographers, may never encounter a case throughout their careers. The case presented here offers a unique perspective on OI, enriched by the familial context spanning four generations.
For this paper, we will be using pseudonyms to refer to the two primary interviewees, "Charles" and "Emma," and their daughter, "Anne."
Family Lineage
Charles’ grandmother is short in stature and was described as a “brittle child” because she experienced a few fractures, mostly affecting her humeri. She lost her adult teeth at 20 and went completely deaf from inner ear bone damage by 25. Her daughter is also a small woman and was described as “fragile” though not to the same degree as her mother. She broke her arm and a rib as a child, but overall had no major red flags to suggest there was an underlying medical condition. She does, however, have slightly blue/grey sclera. The sclera is the whites surrounding the lens of the eye. When this is thin from insufficient collagen, we can see the vasculature behind the eye, producing the classic blue appearance. As an adult, her only health complications are post-menopausal osteoporosis and mild hearing loss, not requiring hearing aids, neither of which can definitively be linked to OI.
“Charles” was born in a remote community with limited prenatal ultrasound access. Because of the lack of access, when his mother went into labour, the hospital was unaware that it was a Frank Breech delivery, creating complications for mother and child. When he was born, his femurs were wrapped around his head due to their bowed nature. He presented with acute angles in both femurs, which were suspected to be healed in-utero fractures.
He was flown to a major city centre, where a team of specialized pediatricians took a detailed family history and conducted a thorough physical exam. Based on his blue sclera, leg deformities, and suspicious family history, he was tentatively clinically diagnosed with OI. Skin biopsies were then obtained from Charles, his mother, and grandmother and the results confirmed COLI1A1 gene mutation with type cast 4 OI, finally diagnosing all three generations.
Because of his bowed femurs, Charles underwent his first major surgery when he was 2 months old. Surgeons performed a double femur osteotomy to attempt to straighten his legs. This was followed by 6 weeks of traction and then 6-8 weeks of a full-body cast. His first unintentional fracture was when he was 2 years old.

As he matured, the growth of Charles' long bones and spine lagged behind, resulting in skeletal malformation. To help with development and structural stability, he would be flown to Montreal to have his long bones surgically fractured and reinforced with rods. The first such surgery was at 7 years old, but he had many throughout his growing stages. When he was 16, he had spinal surgery to correct severe scoliosis, where every vertebrae was fractured and then his spine was straightened and reinforced with a rod. Growing up, Charles sustained many fractures, mainly of the lower limbs, though he broke his thumb and some ribs. He walks with leg braces and a crutch because one of his hips has coxa vara, where the femoral neck and shaft angle are too narrow, causing leg disproportionality.
Because there was not a lot of awareness of the disease and very few treatments when he was a child, his parents were told to be careful, but otherwise, he was free to explore life in whatever manner he deemed safe. He is big on outdoor adventures, including camping, hiking, ice fishing, SUP-ing, and cycling, just with safety and mobility precautions. He does remember his mother becoming hyper-fixated on the health of his bones, making him drink milk exclusively and exploring alternative and Chinese herbal remedies. He describes his childhood as normal but with a few surgeries and fractures. My friend does not identify as “disabled”, but says he has a disability that sometimes requires modifications or forethought.
Because OI is a non-sexed linked, autosomal dominant genetic mutation, there is up to a 75% chance of having an offspring with OI when one of the parents also has the dominant form (OIF, 2024). Due to his lifelong struggle with extreme surgeries, complications from fractures, growth restriction, and lifestyle modifications, he wanted to prevent this from happening to his own children. When it came time to starting his own family, Charles and his wife, who we shall call “Emma”, explored gene selection IVF. They had to create a gene-specific test involving DNA samples from Charles, his mother, and his grandmother. This is only available out of the United States, which led to major costs and transportation delays in their fertility treatments. After several rounds of egg retrievals, they had nine embryos of high enough quality to be genetically tested, of which seven tested positive for COL1A1 gene mutation, one had Turner’s Syndrome, and only one was genetically normative. The single embryo transplant did not result in a pregnancy. They became pregnant naturally with a beautiful little baby, who tested positive via chorionic villi sampling for the same COL1A1 gene mutations as her dad.
Collagen
To understand Osteogenesis Imperfecta, we must first understand collagen. Collagen is a complex protein comprising one alpha2 polypeptide chain and two alpha1 polypeptide chains, which wrap around each other to form a triple helix (Gregersen & Savariraya 2019). These triple helix procollagen chains interweave and overlap, creating a bonded material known as collagen fibrils (Gregersen & Savariraya 2019). This spiral process provides significant strength and stability, preventing injury through tension or pulling mechanisms. This structure avoids injury like climbing or sailing ropes with excellent tensile strength and dynamic elasticity. Due to these two distinct properties, collagen can be found in the fibrous components of connective tissues, namely tendons, ligaments, fascia, sheaths, bursas, bones, cartilage, corneas, and skin (Lindhal et al., 2013).

There are approximately 15 types of collagen, with Type 1 being the most abundant, comprising up to 90% of all collagens (Gregersen Savariraya 2019). These fibres need to interweave and coil on themselves in specific ways to ensure connective tissues’ proper function and integrity. When this order is lost, we get musculoskeletal conditions. Osteogenesis Imperfecta is caused by gene mutations that affect the Type 1 collagen-producing genes, compromising the quality or quantity of collagen (Forlinio et al., 2012).
Understanding the role of collagen helps to understand the physical manifestation of OI. How the disease presents itself depends on the quality and quantity of collagen (Charnas & Marini, 1993; Forlinio et al, 2012). While there are different classification systems, most individuals with OI, regardless of the genetic underlying cause, will share certain features. Those with OI are often described as “fragile” due to the principal characteristic of easily broken bones. As a result, they are usually subject to skeletal deformities and growth restriction. Other common features include blue/grey sclera, compromised tooth integrity, especially among adult teeth, resulting in discoloured, weak teeth prone to cavities and damage, and hearing loss from inner ear bone weakness. Due to its wide clinical presentation, OI used to be characterized primarily by its phenotypical presentation. Many sonographers may know the Types 1-4 classification systems.
Classification and Presentation of Osteogenesis Imperfecta
Type 1 is the mildest phenotypical presentation. Individuals will often have a combination of the traditional OI features but to a lesser degree. Since this is the mildest form, individuals rarely suffer from bone deformities and often have close to normal stature. Type 1 is an outlier; a genetic mutation does not cause it but results from a null COLIA1 allele, meaning it was sufficient quality of collagen but insufficient quantity. This can be autosomal recessive or a random genetic mutation.
Type 2 is the result of deformities of the COLIA2 or COL71A1 genes (Weaver et al, 2021). It may be the form sonographers are most familiar with, as it can be detected prenatally with devastating findings. Fetuses can present with bowed long bones or in utero fractures, reduced bone ossification, and a large, soft cranium (Weaver et al, 2021). These pregnancies are rarely viable due to cardiopulmonary constriction and the multitude of lethal fractures in utero and upon delivery (Deguchi, 2021).
Type 3 is a progressive form that affects both COL1A1/COL1A2. It is the most severe, non-lethal form of OI (Weaver et al, 2021). Those with Type 3 OI can sustain hundreds of fractures throughout their lifespan, resulting in skeletal deformities, compromised bone growth and thus short stature, scoliosis or vertebral compression, frontal bossing, and triangular faces. This progressive deforming subtype often reduces the quality and quantity of life (Deguchi, 2021).
Type 4 is a moderate phenotypical presentation which is often clinically used to fill in the grey area between the two extremes of Type 1 and Type 3 (severe but non-lethal) OI diagnoses. As with Type 2, intrauterine fractures, bowing of long bones, growth restrictions, and demineralization of bones may be seen, but to a significantly lesser degree than Type 2. Types 3 and 4 are rarely fatal to fetuses or neonates.

Type 1 is insufficient collagen due to a null gene mutation. Types 2-4 are caused by mutations affecting the collagen structure). These mutations can alter the size, length, helical pattern, folding, gene sequencing, and matrix organization of Type 1 collagen, which accounts for the wide spectrum of clinical features of OI.
Recessive OI
A small percentage of cases are not caused by dominant gene mutations. 5 gene mutations have been associated with causing recessive OI, which accounts for 2-5% of OI cases (Forlinio et al, 2012; Bodian et al, 2009). Instead of targeting Type 1 collagen structure and quality, recessive OI alters enzyme, protein and amino acid production, thus having an indirect impact on bone growth and integrity, so there is an insufficient quantity of otherwise structurally sound collagen, as seen in Type 1 dominant OI (Forlinio et al. 2012).

The traditional classification of Osteogenesis Imperfecta was initially based on observable characteristics in individuals, predating our current understanding of the underlying genetics. As early genetics were discovered, the strange hybrid of the above four dominant types, and three recessive types was created. Since then, research has identified over 1500 dominant and autosomal recessive mutations in the COLIA1 or COLIA2 genes, revealing a much wider spectrum of genetic variations (Forlinio et al, 2012). This newfound knowledge has rendered the traditional classification system obsolete (Allmann Updyke & Welsh, 2024).
Traditional classifications are limited because they exclude individuals with brittle bones and related long-term effects due to non-Type 1 collagen genetic factors or those not following an autosomal recessive pattern. This underscores the need for a more sophisticated and inclusive categorization system (Allmann Updyke & Welsh, 2024). However, the complexity of genetic causes, overlapping clinical features, and the condition's rarity have prevented establishing a universally accepted classification system. As a result, ongoing efforts focus on developing hybrid systems that consider both phenotype and genetic expression. For example, Charles' mother has a COL1A1 genetic mutation. Still, she presents with such mild symptoms that she identifies as Type 1, although genetically, she aligns more closely with a mild form of Type 4.
Testing Options
Sonographically, Osteogenesis imperfecta may first appear like a general skeletal dysplasia, so more tests are needed to confirm it in the fetus. Ultrasound alone has an accuracy of about 65% for Type II and Type III (Weaver et al., 2021). This accuracy can be improved with prenatal genetic testing or a low-dose CT scan after 26 weeks of pregnancy to check the whole fetal skeleton, and it can be confirmed definitively with invasive or non-invasive tests (Deguchi et al, 2021).
It benefits families and medical care providers when a diagnosis can be achieved early in pregnancy. This can be done through Non-Invasive Prenatal Testing (NIPT). NIPT uses circulating cell-free fetal DNA from maternal blood for genetic testing (Dhallan et al, 2007). It is particularly good for autosomal dominant disorders but only captures OI caused by COL1A1 or COL1A2 gene mutations (Deguchi et al, 2021). This means there is a certain degree of false-negative in the event of autosomal recessive OI, gene mutations in the COL71A1, or a rare mutation affecting other collagen-producing genes.
Invasive Testing
Despite a positive result on NIPT, families continuing pregnancies are still encouraged to confirm with early ultrasound screening and either chorionic villus sampling (CVS) or amniocentesis for better genetic and natal management (Deguchi et al, 2021). Amniocentesis and CVS are the gold standards for diagnosing aneuploidy and genetic diseases in utero (Deguchi et al, 2021). The drawbacks to these invasive procedures are parental stress and physical discomfort of the pregnant person during the procedure. Historically, there was a concern that the risk of miscarriage might be higher, but a recent 2019 review by Salomon et al. has shown that this isn't a strong indicator. Despite this, how people perceive the risk could still affect how likely patients are to choose the procedure.

Charles and Emma opted to forgo the NIPT because they knew they would have to have a CVS regardless of the NIPT results because of the high degree of heritability, their seven positive embryos during their IVF journey, and their natural conception.
Post-natal diagnosis depends on the phenotypical presentation, clinical findings, family history, and radiographic findings (Deguchi et al., 2021). Gene therapy can be provided to determine the underlying cause of OI in an individual or family setting and can be used for fertility counselling either pre- or post-conception.
Sonographic Findings
Ultrasound is the main modality for the in-utero diagnosis of OI based on non-specific sonographic findings (Weaver et al, 2021). It is most useful for initially detecting suspected skeletal dysplasia, monitoring cases of known OI, or tentatively diagnosing Type II OI (which is the most lethal form) (Thompson, 1993).

As Anna has Type 4 OI, she had in-utero findings that were monitored by the Maternal Fetal Medicine team. She had demineralization of bones, healing fractures in the femur, bowing of the femurs (but not humeri), and was measuring small for gestational age. It is important to note that she measured small because her long bones are bowed, underestimating their true length, and both her parents are under 5 feet, meaning her interval growth was appropriate but low against the population average.
At each serial ultrasound appointment, the sonographer evaluated existing fractures during and looked for new breaks, particularly in the long bones. It is, therefore, so important to really zoom up the images so that the area of focus takes up 80-90% of the screen, as this can be extremely subtle. The sonographer would also monitor ribs to make sure they were structurally sound and growing properly, as pulmonary hypoplasia is a serious complication.
Sonographic features include:



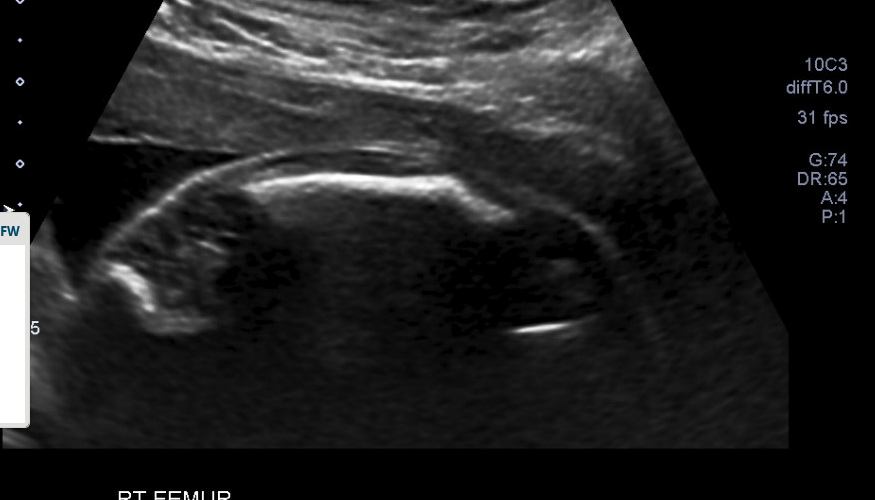





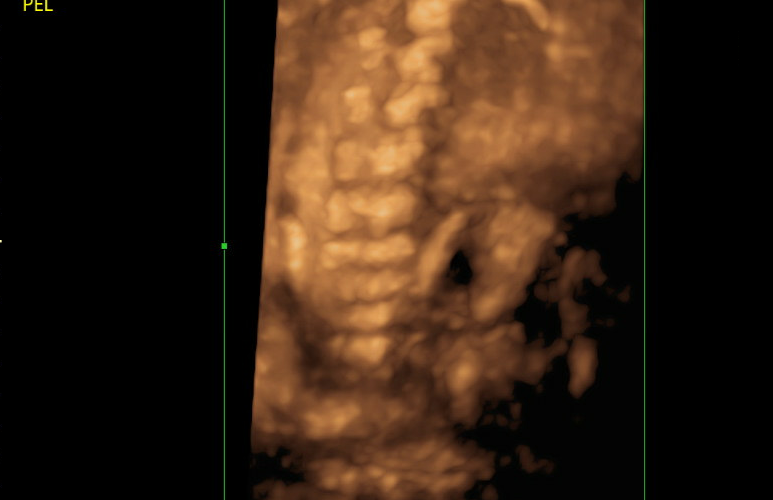


Intrauterine fractures
Bowed long bones
Short, straight ribs
Demineralization of bones
Intrauterine scoliosis
Small for gestational age
Macrocrania or frontal bossing
Type 2 sonographic features demonstrating low survival expectancy are
o Fetal hydrops
o Pulmonary hyperplasia due to thoracic hypoplasia
o Cloverleaf skull (Kimura et al, 2015)






Images retrieved from https://doi.org/10.1007/s10396-015-0645-1 and https://doi.org/10.1515/crpm-2015-0059
Special measurement ratios can be applied to fetuses with skeletal dysplasias, not only OI, to help determine the likelihood of fetal survival (Weaver et al, 2021). This is best evaluated with regular interval obstetrical scans prior to birth.
In cases of skeletal dysplasia:

Femur length ratio to biparietal diameter is lower than normal, ranging from 0.3 to 0.36 instead of the usual 0.44 (Murrin et al., 2022)
Femur length to foot length is between 0.76 and 0.94, compared to the normal range of 0.94 to 1.1 (Murrin et al., 2022)
Femur length to abdominal circumference ratio less than 0.16 is lethal in 92-96% of cases, with worse fetal outcomes in the combined presence of polyhydramnios (Murrin et al., 2022)
Chest circumference to abdominal circumference ratio of less than 0.6 is lethal in 86% of cases (Murrin et al., 2022)
Sonographic Findings Frank Breach
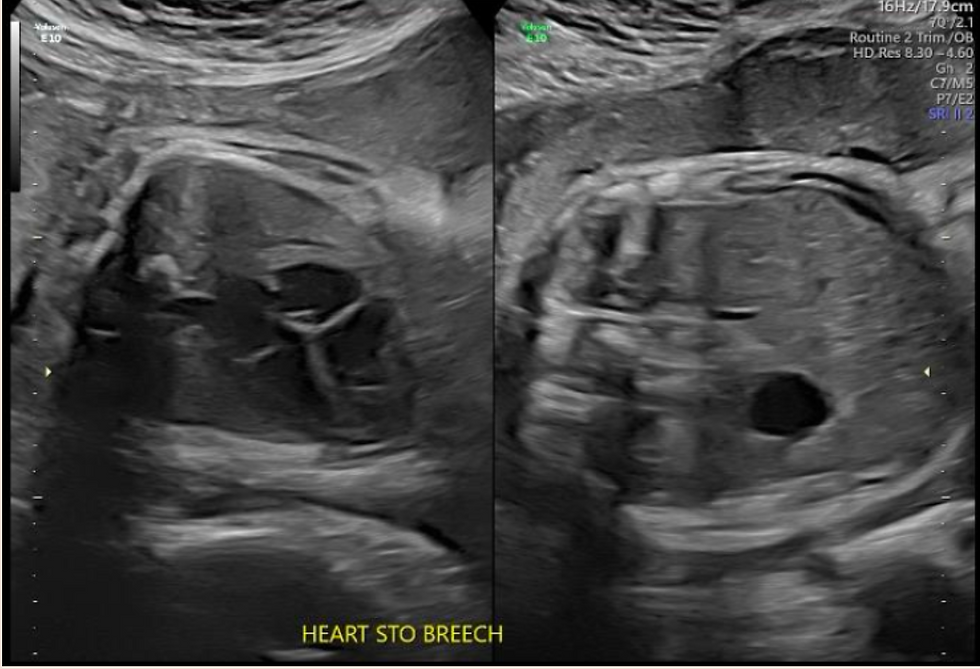
From a delivery perspective, it is important to note that breech lie is significantly higher among OI fetuses, which may impact delivery. Interestingly, multiple studies are showing that there is no significant difference between cesarean versus vaginal delivery in terms of non-lethal fetal fractures at the time of delivery. In other words, cesarean section does not significantly reduce the risk of a fracture at birth (Bellus et al, 2016; Cubert et al, 2001). My friend opted for a c-section for a couple of reasons. The first is that, if all risks were equal between vaginal and c-section delivery, she felt that with a c-section, the OB/GYN [jO1] would have at least some control over external factors. The second reason is that the baby was presenting Frank Breach, and therefore, a c-section was recommended regardless of the underlying diagnosis.

Baby “Anne” was delivered at 35w6d because she had not had sufficient interval growth on serial ultrasounds, a common side effect of the blood pressure medication Emma was on to help control gestational hypertension. Since delivery, Anne’s growth has been good, although she is still on the small end; that could be genetics beyond OI, as both her parents are under 5ft. The baby was also born with healing left femoral fractures and bilateral bowed femurs. Unlike her father, who had major corrective surgery for his bowed legs, Anne was given an x-ray after delivery to evaluate the femoral fractures and was allowed to heal without intervention. Regular pediatric visits have demonstrated good healing of the fractures
and no excessive bowing of the legs.
Advice for General Obstetrical and Maternal Fetal Medicine Sonographers
Continuity of Care
Emma had her MFM ultrasounds done by two different clinics in the city. At the first location, they worked with a few different sonographers. This meant that the burden of explanation and education kept falling on the parents at each ultrasound appointment, which was emotionally exhausting. They described themselves as “going on the defence” to start off the appointment with each new staff to set expectations and tone of the appointment.
The second location maintained the same OB/GYN and sonographer throughout the pregnancy. As a result, a deep and personal relationship grew between the parents and their medical team. This meant that the parents could attend the ultrasound with excitement and ease.
Emma and Charles recommend doing everything in your institution’s power to maintain continuity of care with as few staff members as possible. If your department cannot provide the same sonographer for the family, then use office communication tools to inform the next sonographer about the family’s communication requests. For example, use in-house communication systems to indicate parents’ wishes, knowledge, and wishes for interactions with sonographers In this particular case, Charles wanted his own diagnosis to be better communicated between staff members to reinforce their understanding of the condition and that he is open to being asked about OI.
Keep the Messaging Simple
Families know there is an abnormality, but ultimately, the parents want to know if the baby is doing well or not. An abnormal pregnancy is still a pregnancy to be celebrated and enjoyed. It does not need to be defined by the abnormality alone.
Allow Families to Be in the Moment
So often, families have negative experiences because the medical team assumes that an abnormality is a bad thing or something to be pitied or tiptoed around. Be factual about the condition and just as exuberant about everything else as you would a typical obstetrical scan. The parents are already mentally and logistically preparing for their new life, so make this small moment as positive and impactful as possible.
Safe Space to Process Information
Remember, too, that just because it is routine for us does not mean it is routine for the family; give them grace and time to process new or confusing information and find somewhere private and respectful for them to gather their thoughts.
Aging with OI
Life expectancy and quality of life in individuals with Osteogenesis Imperfecta vary significantly depending on the severity of the condition. Common issues include osteoporosis and declining mobility. Repeated skeletal damage may cause deformities or scoliosis, compressing internal organs, leading to organ failure or pulmonary disease. Additionally, collagen abnormalities in the arteries and veins may result in aneurysms or aortic root dilations, contributing to premature death.
Non-physical effects of Osteogenesis Imperfecta also impact individuals’ aging experiences. Many people can feel isolated, infantilized, or socially excluded because of their physical differences or mobility aids (Hill et al., 2021). It can be mentally exhausting being in a regular state of caution to prevent injury or in a state of recovery from injury (Hill et al., 2021). Even simple activities or procedures carry significant risks; Charles broke his jaw while eating a stale pizza crust five days after getting his wisdom teeth removed. The thought was that there was a minor fracture that occurred at the time of the coronectomy, weakening the mandible and resulting in significant complications.
Treatment
The most prominent “treatment” focuses on preventative physical therapy as early and consistently as possible among those with OI (Botor et al., 2021). This maintains gross motor function, promotes bone strength and integrity, and encourages independence for as long as possible. Thereupon, most treatments are reactionary, such as pain management, mobility aids (wheelchairs, braces, hearing aids), dental surgeries, orthopedic surgery, and rehabilitation (Botor et al., 2021).

Currently, no dedicated treatments for OI exist (Dimulescu et al., 2024). To improve bone integrity, patients with OI have been treated with bisphosphates, which is the same medical classification as seen in the treatment of osteoporosis (Martin & Seeman, 2008). The theory is that reducing the bone turnover rate improves mineralization and thus bone structural integrity and hopefully reduces the number of fractures an individual will get throughout their lifespan. Since most complications from OI are the result of multiple fractures, this will improve morbidity and mortality rates among older individuals and promote autonomy.
A review from 2024 showed over 8 different forms of indirect treatments for OI at the delivery or clinical trial phase (Dimulescu et al., 2024). Sclerostin Inhibitors are monoclonal antibodies that inhibit a small protein that is active in the remodelling process of bones (Dimules, 2024). Decreasing the remodelling rate of bones increases the amount of collagen in
bones, hypothetically leading to stronger bones. Just like bisphosphonate, sclerostin inhibitors are osteoporosis treatments that are being retrofitted to attempt to help in other diseases with compromised bone mineralization. Teriparatide is another osteoporosis medication targeting the parathyroid hormone, stimulating bone mineralization, density, and strength currently undergoing clinical trials (Leali et al., 2017). It increases osteoblast longevity and quantity, thus increasing calcium levels and promoting bone density (Dimulescu et al. 2024).
Charles and Emma chose a modern form of bisphosphonate therapy called zoledronic acid. Their daughter had her first IV treatment when she was five days old and will have treatments every three months until she is two years old. At that point, it will go to every six months, with routine bone mineral density surveillance to determine how long she will get this therapy.
As a baby, Anne has been seeing a physical therapist on a routine basis. She initially went for treatment for torticollis, which is common with breached babies. They have continued working with this physical therapist monthly to help with gross motor development expectations and will continue to help support Anne with weight-bearing activities as she grows.
Gene therapy
Gene selection or gene counselling pre- or post-conception offers options to families with a high risk of conceiving a fetus with OI. This poses ethical and financial pressure on those seeking counselling and who choose to move forward with gene selection IVF. It is also logistically challenging as there are very few genetic testing facilities in North America equipped to test embryos for OI, and often an individual test must be generated at the expense of those seeking treatment.
Gene therapy is another promising option since 85-90% of cases of OI are the result of gene mutations in the COL1A1 and COL1A2 genes (Lindahl et al. 2013). siRNA gene silencing may help manage most cases by silencing abnormal gene presentation on these two primary alleles (Ljungreen et al., 2011). This is preliminary but shows promise in future application of direct OI treatment and prevention. It does not address null allele or recessive gene mutations.
Life Beyond a Diagnosis
When I sat down with my friends to ask what they wanted me to share on their behalf, they looked at each other and nodded as though they had been expecting this question. They then turned to me, and Charles said, “Having OI is not as bad as the clinical opinion. The challenges would be similar to any other real-life challenge, like going through a parental divorce or being picked on in school. It’s not a massive hindrance. At times I felt left out, but it’s hard to tell if that was because of OI or because kids can suck”.
He reiterated that he had so many medical interactions starting at such a young age that it just felt like a normal part of his life. Neither the medical interventions nor living with OI are as traumatic as people think it would be. He also says that he got used to breaking bones.
The clinical diagnosis of OI does not reflect the lived experience. There is a wide range of condition expressions, from mild, like his mother, to more severe forms than Charles. Most medical staff are familiar with the extreme versions and thus feel as though the diagnosis is more detrimental than it can be. Medical staff at all support stages need more education about the spectrum of OI. Charles was never held back from experiencing life to its fullest. He may have needed some modifications, but he still does it all.
Raising a child with OI
Since having a daughter with OI, Charles has had some profound conversations with his parents about what it is like raising a child with this condition. He states he has many more opportunities and resources than his parents ever had. In addition to having first-hand experience with the condition, medical awareness and treatment options have improved significantly since he was a child. He knows what questions to ask and how to advocate for his daughter's medical needs and safety. Emma is an early childhood educator and advocator for members of the deaf and hard-of-hearing community. She knows what resources are available to students and how to advocate for her daughter. Similarly, she knows when they do not need to take advantage of accommodations and can encourage autonomy and independence.
Despite this, Charles says, “It is still scary because it is your child.” He watches his daughter interact with the world and sees things differently from his wife. One poignant example he provides is when he sees his daughter standing with a leg by the baby gate. Emma will watch to ensure Anne does not fall or the gate does not drop on her. Charles, on the other hand, will go and adjust the angle of her leg because when he was a baby, he broke his femur by getting it twisted up in the rods.
Ultimately, both parents agree that, like Charles’ parents before him, he will not limit Anne’s life or subject her to a lifetime of fear of injury. They prioritize experiencing life to the fullest and pushing boundaries for their daughter. Both parents believe that injuries are a normal part of childhood, regardless of any underlying diagnosis, and that, ultimately, the diagnosis does not define her.
References
Allman Updyke, E. & Welsh, E. (2024, January 2). Osteogenesis Imperfecta: All Bones About It. This Podcast Will Kill You. https://thispodcastwillkillyou.com/2024/01/02/ep-132-osteogenesis-imperfecta-all-bones-about-it/
Bellur, S., Jain, M., Cuthbertson, D., Krakow, D., Shapiro, J. R., Steiner, R. D., ... & Nagamani, S. C. (2016). Cesarean delivery is not associated with decreased at-birth fracture rates in osteogenesis imperfecta. Genetics in Medicine, 18(6), 570-576.
Bodian DL, et al. Mutation and polymorphism spectrum in osteogenesis imperfecta type II: implications for genotype-phenotype relationships. Hum Mol Genet. 2009;18:463–471. https://doi.org/10.1093/hmg/ddp083
Botor M, Fus-Kujawa A, Uroczynska M, Stepien KL, Galicka A, Gawron K, Sieron AL. Osteogenesis Imperfecta: Current and Prospective Therapies. Biomolecules. 2021; 11(10):1493. https://doi.org/10.3390/biom11101493
Charnas, L.R. & Marini, F.C. (1993). Communicating Hydrocephalus, basilar invagination and other neurologic features in osteogenesis imperfecta. Neurology Journals 34(12). https://doi.org/10.1212/WNL.43.12.2603
Cubert, R., Cheng, E. Y., Mack, S., Pepin, M. G., & Byers, P. H. (2001). Osteogenesis imperfecta: mode of delivery and neonatal outcome. Obstetrics & Gynecology, 97(1), 66-69.
Deguchi M, Tsuji S, Katsura D, Kasahara K, Kimura F, Murakami T. (2021). Current Overview of Osteogenesis Imperfecta. Medicina. 57(5):464. https://doi.org/10.3390/medicina57050464
Devogelaer, Jean-Pierre, et al. “Radiological manifestations of bisphosphonate treatment with APD in a child suffering from osteogenesis imperfecta.” Skeletal radiology 16 (1987): 360-363
Dhallan, R.; Guo, X.; Emche, S.; Damewood, M.; Bayliss, P.; Cronin, M.; Barry, J.; Betz, J.; Franz, K.; Gold, K.; et al. A non-invasive test for prenatal diagnosis based on fetal DNA present in maternal blood: A preliminary study. Lancet 2007, 369, 474–481
Dinulescu A, Păsărică A-S, Carp M, Dușcă A, Dijmărescu I, Pavelescu ML, Păcurar D, Ulici A. New Perspectives of Therapies in Osteogenesis Imperfecta—A Literature Review. Journal of Clinical Medicine. 2024; 13(4):1065. https://doi.org/10.3390/jcm13041065
Gregersen, P.A., & Savariraya, R. (2019). Type II Collagen Disorders Overview. Europe PMC. PMID:
31021589.
Hill, M., Hammond, J., Sharmin, M., et al. (2021). Living with Osteogenesis Imperfecta: A Qualitative Study Exploring Experiences and Psychosocial Impact from the Perspective of Patients, Parents, and Professionals. Disability and Health Journal (15(1). https://doi.org/10.1016/j.dhjo.2021.101168
Hoyer-Kuhn, H.; Netzer, C.; Semler, O. Osteogenesis Imperfecta: Pathophysiology and Treatment. Wien. Med. Wochenschr. 2015, 165, 278–284.
Interview with Parents by pseudonyms Charles, Emma, and Anne. 2024.
Kimura, I., Araki, R., Yoshizato, T. et al. A case of fetal osteogenesis imperfecta type 2A: longitudinal observation of natural course in utero and pitfalls for prenatal ultrasound diagnosis. J Med Ultrasonics 42, 565–570 (2015). https://doi.org/10.1007/s10396-015-0645-1
Leali, P.T.; Balsano, M.; Maestretti, G.; Brusoni, M.; Amorese, V.; Ciurlia, E.; Andreozzi, M.; Caggiari, G.; Doria, C. Efficacy of teriparatide vs neridronate in adults with osteogenesis imperfecta type I: A prospective randomized international clinical study. Clin. Cases Miner. Bone Metab. 2017, 14, 153–156
Lindahl, K., Kindmark, A., Laxman, N. et al (2013). Allele Dependent Silencing of Collagen Type I Using Small Interfering RNAs Targetting 3’UTR Indels – a Novel Therapeutic Approach in Osteogenesis Imperfecta. International Journal of Medical Sciences, 10(10): 1333-1343. doi: 10.7150/ijms.5774
Ljunggren, Ö.; Lindahl, K.; Rubin, C.-J.; Kindmark, A. Allele-Specific Gene Silencing in Osteogenesis Imperfecta. Endocr. Dev. 2011, 21, 85–90
Martin, T.J.; Seeman, E. Bone remodelling: Its local regulation and the emergence of bone fragility. Best Pract. Res. Clin. Endocrinol. Metab. 2008, 22, 701–722.
Murrin, E.M., Nelsen, G., Apostolakis-Kyrus, K. et al. (2022). Evaluation of First Trimester Ultrasound Fetal Biometry Ratios Femur Length/Biparietal Diameter, Femur Length/Abdominal Circumference and Femur Length / Foot for the Screening of Skeletal Dysplasia. Obstetrics and Gynecology, 43(7): 919-928. https://doi.org/10.1002/pd.6295
Novak, L., Steinberger, D., Wilhelm, A. & Bahlmann, F. (2016). Osteogenesis Imperfecta type II with the variant c.4237G>A (p.Asp1413Asn) in COL1A1 in a dichorionic, diamniotic twin pregnancy. Case Reports in Perinatal Medicine, 5(1), 45-48. https://doi.org/10.1515/crpm-2015-0059
Parilla, B.V., Leeth, E.A., Kambich, M.P. et al. (2003). Antenatal Detection of Skeletal Dysplasia. Journal of Ultrasound in Medicine 22(3): 255-258. https://doi.org/10.7863/jum.2003.22.3.255
Osteogenesis Imperfecta Foundation (OIF), 2024. https://oif.org/informationcenter/about-oi/
Salomon, L.J., Sotiriadis, A., Wulff, C.B. et al (2019). Risk of Miscarriage Following Amniocentesis of Chorionic Villus Samplling: Systematic Review of Literature and Updated meta-Analysis. Ultrasound Obstetrics and Gynecology, 54: 4420451. https://doi.org/10.1002/uog.20353
Thompson, E.M. (1993). Non-Invasive Prenatal Diagnosis of Osteogenesis Imperfecta. American Journal of Medical Genetics, 45(2): 201-206. https://doi.org/10.1002/ajmg.1320450210
Van Dijk, F.S., Pals, G., Van Rijn, R.R. et al (2010). Classification of Osteogenesis Imperfecta Revisited. European Journal of Medical Genetics. 53(1): 1-5. https://doi.org/10.1016/j.ejmg.2009.10.007
Weaver, J.S., Revels, JW., Elifritz, J.M. et al (2021). Clinical Manifestations and Medical Imaging of
Osteogenesis Imperfecta: Fetal Through Adulthood. Acta Medica Academica, 50(2). DOI: 10.5644/ama2006-124.343




Comments